ADHD
ADHD vigilance regulation sleep
Below we will cover in more depth some relevant sub-groups in ADHD with relevance to understanding ADHD and also with relevance to treatment outcome, as well as recommendations for future research. The results below have already resulted in new and more recent insights about the role of sleep, the biological clock (circadian system) and ADHD, also see the section on ‘Sleep and ADHD’.
Impaired vigilance regulation
The main finding from the ADHD studies is that a specific sub-group of ADHD patients was found who can be characterized by a lower vigilance and more unstable vigilance regulation who are responsive to stimulant medication. This is consistent with a review on sleep problems in ADHD where the most consistent finding across studies was that of daytime excessive sleepiness (Cohen-Zion & Ancoli-Israel, 2004). In earlier research it was reported that ADHD children characterized by frontal alpha and frontal theta – reflective of lower vigilance stages – were the best responders to stimulant medication (Arns et al., 2008). Furthermore, in the research by Sander et al. (2010) further indications were found that ADHD patients were characterized by a lower vigilance as well as a more unstable vigilance regulation. There also was a tendency for the ADHD patients with the lowest EEG vigilance to perform worst on a CPT test, reflective of inattention and impulsivity, and these patients tended to respond better to medication as well. These findings are in line with the EEG Vigilance model originally developed by Bente (Bente, 1964) and recently further developed by Hegerl (Hegerl et al., 2010), and are hypothesized to explain the ADHD symptoms and subtypes. As can be seen in figure 1 below, a trait-like unstable vigilance regulation explains the cognitive deficits characteristic for ADHD and ADD such as impaired sustained attention. Furthermore, the vigilance autostabilization behavior explains the hyperactivity aspect of ADHD as an attempt to upregulate vigilance.
ADHD vigilance regulation sleep
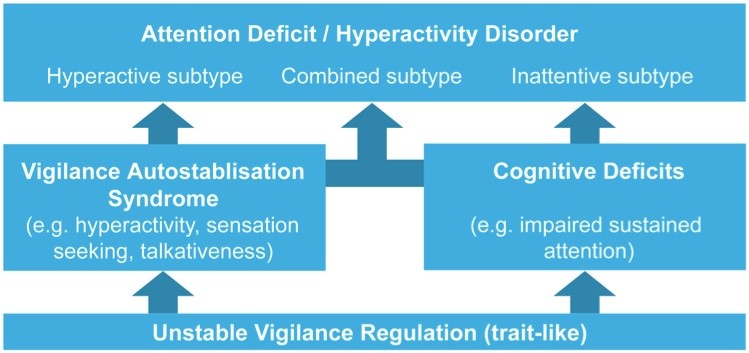
Figure 1:This figure provides an overview of the relation between an unstable vigilance regulation and the behavioral symptoms of ADHD (adapted from Hegerl et al. 2009).
The EEG vigilance model is more focused on examining the temporal dynamics of the EEG rather than focusing on the EEG activity averaged across time. The different vigilance stages are reflective of the same underlying process (vigilance) and hence changes in alpha or theta power alone are of little value in establishing a relationship with behavior. The EEG Vigilance model explains the relationship between EEG phenomena and behavior by means of vigilance regulation. The interesting aspect of this model is that this relationship between behavior and EEG is not a linear relationship, explaining why in chapter 4 no consistent relationship between EEG power measures and neuropsychological performance was found. The following example illustrates this further.
After a tiring day the EEG vigilance regulation of a healthy person will become unstable and demonstrate more of the lower vigilance stages. This has a very well known EEG signature often referred to as ‘fatigue’ or ‘drowsiness’ expressed as alpha anteriorisation (Broughton & Hasan, 1995; Connemann et al., 2005; De Gennaro et al., 2001; De Gennaro et al., 2004; De Gennaro et al., 2005; Pivik & Harman, 1995) and increased frontal slow waves (Strijkstra et al., 2003; Tanaka et al., 1996; Tanaka et al., 1997), in the EEG vigilance model referred to as stage A2-A3 and B2-B3 respectively. In young children we all know the example of the hyperactive, ‘high-spirited’ behavior in over-tired children, as a clear example of vigilance autostabilization behavior (i.e. keeping himself awake). A healthy adult displaying this type of EEG – assuming he is at home and it is almost bedtime – will subjectively feel sleepy and decide to go to bed (i.e. the adult will decide to ‘withdraw’ from the environment and seek an environment with low external stimulation and increase the chance to fall asleep).
However, when this same healthy adult is driving a car and exhibits the same reduced EEG Vigilance and is drowsy, he will turn up the volume of the music… open the window… turn-down the air-conditioning… close the window…and so on. Hence the healthy adult will exhibit autostabilization behavior in order to keep himself awake. Furthermore, when the car in front of him will unexpectedly brake, he will be more likely to respond slowly (impaired sustained attention) and the likelihood of a car accident increases due to this reduced vigilance or fatigue (Miller, 1995).
ADHD vigilance regulation sleep
This example nicely illustrates that the same EEG state – depending on the environment – can result in completely different behavior (sleep vs. ‘hyperactive’ behavior), thereby explaining why the relationship between EEG and behavior is not a linear relationship, but rather a ‘binary relationship’. Furthermore, as pointed out above, changes in alpha or theta averaged across time provide little information, since both are reflective of the same process, namely vigilance. Therefore, for future studies investigating the relationship between resting EEG and cognition it is recommended to rather employ an overall EEG vigilance metric rather then investigating the averaged spectral content ‘in isolation’ and also investigate this during a task. For example in a future study it is planned to investigate the EEG Vigilance stages in patients with ADHD during a CPT task and investigate if lower vigilance stages preceding a target requiring a response, result more often in an omission to respond, reflective of inattention (false negative errors). Previous studies have investigated the relationship between alpha and theta and cognitive processes and have reported that during a task increased alpha is related to memory load (Jensen & Mazaheri, 2010; Jensen et al., 2002) and increased theta is related to short-term memory (Klimesch, 1996; Osipova et al., 2006). These studies hence reflect task-induced synchronization in the theta and alpha band to be associated with improved mnemonic function. However, studies investigating pre-stimulus EEG power have demonstrated that increased pre-stimulus alpha is associated with a decrease in visual discrimination ability (van Dijk, Schoffelen, Oostenveld, & Jensen, 2008), with false alarms in a Go-NoGo task (Mazaheri, Nieuwenhuis, van Dijk, & Jensen, 2009) and attentional lapses (O’Connell et al., 2009). This latter study found that alpha started to increase already up to 20 s. before a missed target (O’Connell et al., 2009). Furthermore, Romani et al (1988) specifically investigated if vigilance, classified using the EEG, was related to subsequent ERP amplitudes and latencies and found that lower pre-stimulus vigilance levels (increased theta and delta) were associated with subsequent slower N100 latencies. They also found indications that subjects with a more unstable vigilance (greater fluctuations) were characterized by slower N100 latencies but also reduced N100 amplitudes with ‘less vigilant’ prestimulus stages (Romani, Callieco, & Cosi, 1988). These results suggest that EEG vigilance stages could indeed explain attentional lapses (contrary to the interpretation by Van Dijk et al. (2008)). However, all these studies were conducted in healthy volunteers and all investigated average EEG power in a specific frequency band, whereas as pointed out above one should rather investigate the EEG vigilance level where anterior alpha and theta reflect a continuum of decreasing stages of EEG vigilance.
ADHD vigilance regulation sleep
Sleep problems as the core pathophysiology of the vigilance sub-group in ADHD
ADHD vigilance regulation sleep
The reduced EEG Vigilance described in the example above can be observed when driving a car very late at night while being fatigued, but can also be caused by sleep deprivation or impairments in vigilance regulation and maintenance. As explained in Hegerl et al., (2010): ‘…sleep deficits or other factors inducing an unstable vigilance trigger an autoregulatory behavioral syndrome with hyperactivity, sensation seeking and distractibility. This behavioral syndrome has the function to stabilize vigilance by creating a highly stimulating environment. In vulnerable individuals, the autoregulatory mechanism overrides the physiological tendency to seek sleep, aggravates the sleep deficits, worsens the vigilance instability and thereby starts a vicious circle…’.
ADHD has been associated with sleepiness, shortened sleep latency (Golan et al., 2004), primary sleep disorders, sleep related movement disorders and parasomnias (Chervin et al., 2002; Konofal, Lecendreux & Cortese, 2010; Walters, Silvestri, Zucconi, Chandrashekariah & Konofal, 2008) and ADHD-like behavior can be induced in children by sleep restriction (Fallone et al., 2001; Golan et al., 2004) and improved by reducing sleep deficits (Dahl et al., 1991). Recently, Van Veen (Van Veen et al., 2010) reported in a sample of adult ADHD patients that 78% had sleep-onset insomnia, confirmed by actigraphy and associated with a delayed nighttime melatonin onset. A similar rate of 73% sleep onset insomnia has been reported in children with ADHD (Van der Heijden, Smits, Van Someren & Gunning, 2005). These data suggest that at least a subgroup of patients with ADHD is characterized by a delayed endogenous circadian phase associated with delayed sleep onset (Van Veen et al., 2010). Several studies have investigated the effects of melatonin as an aid to shift this circadian phase and most have reported clear improvements in sleep onset insomnia (Hoebert, van der Heijden, van Geijlswijk & Smits, 2009; Van der Heijden, Smits, Van Someren, Ridderinkhof & Gunning, 2007), however after 4 weeks treatment no improvements of ADHD symptoms and cognition were reported (Van der Heijden et al., 2007), whereas after long-term treatment improvements of behavior and mood were reported only for those children who were still using melatonin and discontinuation usually resulting in a relapse into sleep onset insomnia (Hoebert et al., 2009). This suggests that in this sub-group, normalizing sleep onset insomnia can be achieved by improving circadian regulation by for example melatonin, albeit with a delayed-onset of effect on ADHD symptoms. The effects of stimulant medication on sleep parameters demonstrates a discrepancy between objective and subjective findings, except for prolonged sleep latency and prolonged onset to first REM cycle (Cohen-Zion & Ancoli-Israel, 2004; Corkum, Tannock, & Moldofsky, 1998) at least demonstrating that stimulant medication does not improve sleep in ADHD. Therefore, the efficacy of stimulant medication has to be sought into the vigilance stabilizing properties during the day.
ADHD vigilance regulation sleep
Earlier research has shown that neurofeedback treatment in ADHD has been well investigated and this treatment is showing promising results in the treatment of ADHD (Arns et al., 2009: Sherlin et al., 2010). The exact working mechanism of this treatment is not well understood, but is thought to be related to the patients’ ability to operantly condition brain oscillating activity (Sherlin et al., 2011). The most often reported protocols used in ADHD are Theta/Beta neurofeedback and SMR neurofeedback and no differential effects of these protocols could be found based on studies providing one of these protocols for a whole group of patients. However, some of our recent work has provided some preliminary evidence that selecting either of these protocols based on the pre-treatment EEG could improve treatment outcomes, most specifically for the domain of inattention.
ADHD vigilance regulation sleep
Several studies have demonstrated that SMR neurofeedback results in increased sleep spindle density during sleep (Hoedlmoser et al., 2008; Sterman et al., 1970) and improves sleep (Cortoos et al., 2010; Hoedlmoser et al., 2008). This suggests that this type of neurofeedback specifically targets the sleep problems, which in turn results in vigilance stabilization (i.e. restore the ‘trait like unstable vigilance regulation’, also see figure 2). In this view then, sleep problems are considered to be the core pathophysiology in ADHD. In turn vigilance stabilization results in improvements of inattention and hyperactivity/impulsivity in ADHD. Interestingly in some of our recent work, it was found that a sub-group who was treated with SMR neurofeedback demonstrated clear neurophysiological changes (increased N200 and P300 amplitude) in oddball ERP and EEG along with clear clinical improvements on core ADHD symptoms. These could be specific effects of this protocol on brain function and subjective reports, although this is still uncertain given the lack of control group. In this regard it is also interesting to note that all patients improved significantly on sleep problems assessed using the Pittsburgh Sleep Quality Inventory (PSQI; unpublished observation), further supporting this notion.
The theta/beta neurofeedback protocol aims at reducing fronto-central theta and increasing beta. In terms of the vigilance model it could be hypothesized that this treatment teaches patients to stabilize their vigilance by decreasing the occurrence of lower vigilance stages, characterized by theta (vigilance stage B2-3) and demonstrating more occurrences of desynchronized beta EEG. More research is required to further substantiate this and investigate the exact working mechanism of theta/beta neurofeedback.
Summarizing, there appears to be a subgroup of ADHD patients in whom there is an impaired vigilance regulation, most likely associated with sleep problems such as sleep onset insomnia. Based on chapter 1, if we consider the frontal slow (B2-3), frontal alpha (A3) and low voltage EEG (B1) as states of lower EEG vigilance, this prevalence is estimated to be 55%. Based on the studies discussed above on sleep onset insomnia this prevalence is estimated at 75% (Van der Heijden et al., 2007; Van Veen et al., 2010). This sleep onset insomnia has been related to a delayed circadian rhythm (Van Veen et al., 2010), and hence normalizing this circadian regulation is hypothesized to improve vigilance regulation and thereby improve ADHD symptoms. These patients respond well to stimulant medication, due to its vigilance stabilizing properties. However, in this view the effects of stimulant medication have to be seen as a symptom suppression not affecting the core pathophysiology of ADHD, but only increasing daytime vigilance. The effects of melatonin – if timed in the appropriate circadian phase – seem to more directly affect the core pathophysiology via its normalizing effect on sleep, albeit with a delayed effect on clinical ADHD symptoms. In line with this delayed onset of effect of melatonin on ADHD symptoms, an interesting hypothesis deserving further study is that maybe neurofeedback can require many fewer sessions, and should be stopped when sleep is normalized. The effects of melatonin however, disappear when it is discontinued. Hence future studies should investigate further how this ADHD sub-group can be identified more reliably by EEG, polysomnography or maybe by sleep parameters such as the ‘dim light melatonin onset’ or DLMO measure reported in several studies (Hoebert et al., 2009; Van der Heijden et al., 2007; Van Veen et al., 2010). Furthermore, future studies should investigate how these sleep problems could be treated more effectively with sustained effects by for example bright light, sleep hygiene or SMR neurofeedback.
The ‘slow individual alpha peak frequency’ sub-group.
ADHD vigilance regulation sleep
The most consistent finding which emerged throughout all studies reported in this thesis is the utility of the slow individual alpha peak frequency (iAPF) sub-group for both ADHD and depression. As pointed out in chapter 1, this measure has often been reported in the older EEG literature and has largely been ignored by many researchers employing QEEG techniques, most likely due to the difficulty of reliably calculating this in an automated fashion. It was found that ADHD patients with a slow iAPF – in contrast to patients with frontal theta – do not respond well to stimulant medication, whereas at baseline they present among the worst performers on a CPT test (inattention and impulsivity errors), illustrating the importance of dissociating these 2 subgroups.
In Figure 2, this is illustrated in more detail. This figure shows the spectral content of ADHD children (red) and data from a control group (black) for both frontal (Fz) and parietal (Pz) locations. The dotted lines reflect the groups with a ‘normal EEG’ and the solid lines show the spectral power of the sub-groups with a ‘Frontal Slow’ (top) or ‘Slowed Alpha peak frequency’ (bottom). As can be seen the spectral content for the Frontal Slow group is increased in the theta frequency range mainly at Fz, as would be expected. However, the ADHD group with the Slowed iAPF at Pz showed an average APF of 7.5 Hz. In the frontal locations this also shows up as an ‘increased theta EEG power’ whereas this obviously is due to the excessive slowing of the iAPF and should be considered slow alpha, not theta.
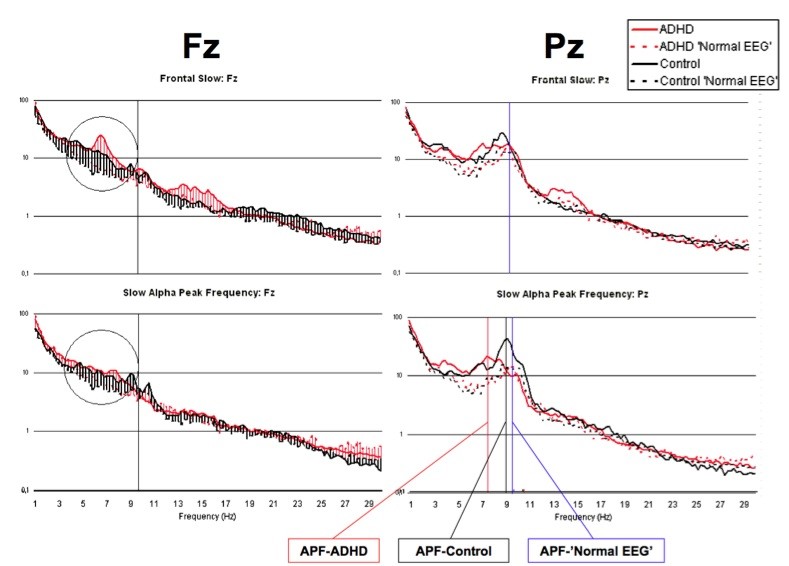
Figure 2: This figure clearly demonstrates that the sub-group with a slowed alpha peak frequency (bottom), present parietally, also exhibit elevated ‘theta EEG Power’ at frontal sites. However, this is not true ‘frontal slow’, but simply the effect of the slowed alpha peak frequency. This demonstrates that a raised theta/beta ratio at least also includes the slow APF sub-group, which neurophysiologically is a different group, as demonstrated with respect to treatment outcome to stimulant medication.
In Lansbergen et al. (2011) it was further demonstrated using a quantitative approach that the often reported increased theta/beta ratio in ADHD actually combines both the frontal slow group (interpreted as the ‘impaired vigilance regulation subgroup’ see above) as well as the slow iAPF subgroup. Therefore, although the theta/beta ratio and the ‘excess theta’ can discriminate a group of children with a DSM-IV diagnosis of ADHD very well from healthy controls (Boutros et al., 2005; Monastra et al., 1999; Snyder & Hall, 2006), this measure is probably not a specific measure since it incorporates different subtypes of ADHD. From a personalized medicine perspective this is not optimal, since these sub-types respond differentially to medication and are hypothesized to have a different underlying pathophysiology.
This also helps explain the contradictory findings between Chabot et al. (1999; 2001) who found that their excess theta group (described as ‘generalized excess of theta absolute and relative power, decreased alpha mean frequency, and frontal theta hypercoherence’) exhibited a lower response to stimulant medication, suggesting they included patients with a low iAPF, versus Clarke et al. (Clarke, Barry, McCarthy & Selikowitz, 2002; Clarke, Barry, McCarthy, Selikowitz & Brown, 2002) and Suffin and Emory (1995) who found that responders to stimulant medication demonstrated increased theta and increased theta/beta ratios.
ADHD vigilance regulation sleep
In some of our recent work it was found that there was no relation between a slow iAPF and the outcome to neurofeedback in ADHD in inattention and impulsivity/hyperactivity. In this sample the prevalence of a slow iAPF might have been too low (iAPF < 8 Hz: N=1 for parietoccipital iAPF and N=6 for frontal iAPF from total N=19) to find a clear relationship between a slow iAPF and treatment outcome on ADHD rating scales. Therefore the conclusion that neurofeedback can be considered an effective treatment for those patients with a slow iAPF who do not respond to stimulant medication is unjustified at this moment. More research with larger samples are required to further investigate that.
Further implications for treatment related to this sub-type will be discussed in the next section on depression, since this sub-type appears as a non-specific predictor for non-response to treatments across disorders.
Excess beta sub-group
There is also clear evidence that in addition to the above discussed 2 sub-groups, a third sub-group exists that is characterized by excess beta or beta spindles, and makes up 13-20% of the ADHD population (Chabot & Serfontein, 1996; Clarke et al., 1998; Clarke et al., 2001b) and which was also observed in chapter 2 and chapter 6. Several studies demonstrated that these patients do respond to stimulant medication (Chabot et al., 1999; Clarke et al., 2003b; Hermens et al., 2005). Relatively little is known about this excess beta group and about beta spindles. The latter are generally observed as a medication effect due to benzodiazepines (Blume, 2006) or barbiturates (Schwartz, Feldstein, Fink, Shapiro & Itil, 1971). Furthermore, Clarke et al. (2001c) reported this ADHD sub-group was more prone to moody behavior and temper tantrums and Barry et al. (2009) reported that the ERP’s of this sub-group differed substantially from ADHD children without excess beta, suggesting a different dysfunctional network explaining their complaints. Interestingly the ERP’s of the excess beta sub-group appear more normal than those of the ADHD sub-group without excess beta.
Originally Gibbs and Gibbs (1950) distinguished two types of predominantly fast EEG, a moderate increased beta, which they termed ‘F1’ and a marked increased beta, which they termed ‘F2’. Records of the F1 type were initially considered as ‘abnormal’ until the 1940’s, whereas since that time Gibbs and Gibbs only considered the F2 type as ‘abnormal’. However, currently electroencephalographers have shown a more lenient philosophy towards fast tracings (From: Niedermeyer & Da Silva (2004) page 161). At this moment the only abnormal EEG pattern in the beta range is the ‘paroxysmal fast activity’ or ‘beta band seizure pattern’, which most often occurs during non-REM sleep, but also during waking (Stern & Engel, 2004). This pattern is quite rare (4 in 3000) and most often seen in Lennox-Gastaut syndrome (Halasz, Janszky, Barcs & Szcs, 2004). Vogel (1970) also described an EEG pattern of ‘occipital slow beta waves’ or also termed ‘quick alpha variants 16-19/sec’ which responds in the same way as alpha to eyes opening and also has a similar topographic distribution. This pattern was only found in 0.6% of a large population of healthy air-force applicants, given it’s very low prevalence and occipital dominance, this subtype is unlikely the explanation of the ‘excess beta’ or ‘beta spindling’ sub-type observed in ADHD. Therefore, the ADHD sub-group with excess beta or beta spindling (assuming the paroxysmal fast activity has been excluded) can neurologically be considered a ‘normal variant’. However, neurophysiologically this can be considered a separate sub-group of ADHD, which does respond to stimulant medication (Chabot et al., 1999; Clarke et al., 2003b; Hermens et al., 2005). More research is required to investigate the exact underlying neurophysiology of this sub-type and if other treatments could more specifically target this excess beta or beta spindling.
Beta spindles: Examples
In figure 3 traces of EEG are shown where (a) is a normal EEG with posterior alpha and frontal desynchronized EEG. (b) shows Synchroneous beta activity frontal, this is rhythmic beta activity, but it does not show a spindling rhythm yet. (c) shows clear beta spindles frontal You can see this is rhythmic beta with a center frequency and it clearly stands out from the back ground activity.
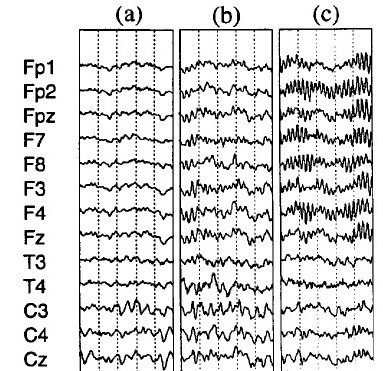
Figure 3 from Clarke et al. (2001)
Figures 4 and 5 show the data of a patient with ‘complex’ ADHD who did not respond to conventional treatments. This client has been treated with Neurofeedback both on downtraining theta frontally and downtraining the beta spindle frequency at Cz in the 24-30 Hz range. Note the quite localized presence of the beta spindles pre-treatment. Pre- and post QEEGs show a clear normalization of the beta spindles. Client also improved clinically during the course of treatment.
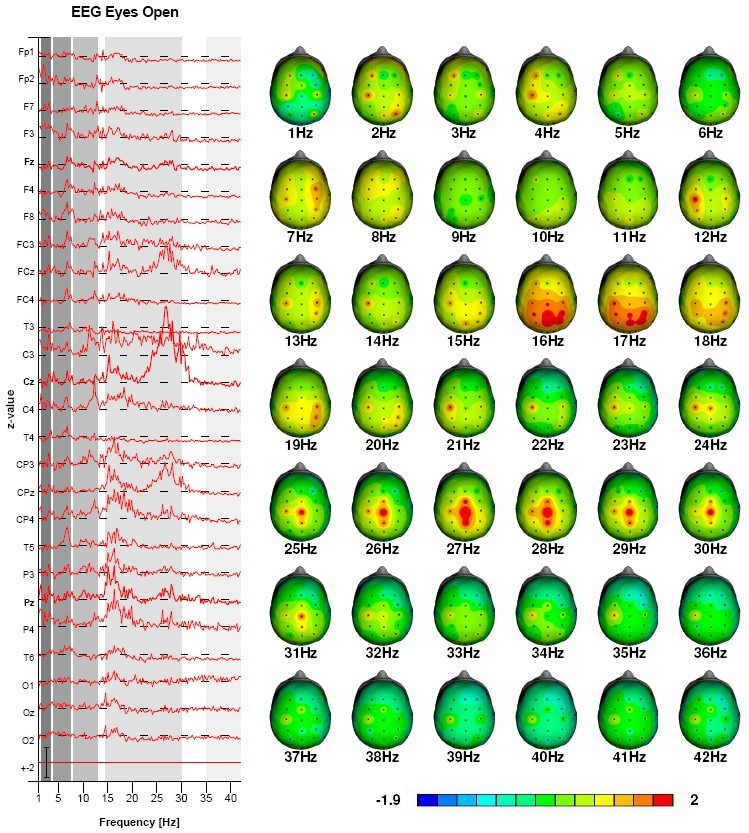
Figure 4: Pre-treatment QEEG: Beta-spindles 25-30 Hz
Figure 5: Post-treatment: Beta-spindles normalized